Research Spotlight: Using Gene Therapy for Pediatric Epilepsy
Manoj Patel, PhD, is a Professor of Anesthesiology at the UVA School of Medicine. The Manoj Patel Lab focuses on sodium channels and epilepsy. By understanding the cellular mechanisms that initiate seizures in SCN8A epileptic encephalopathy, they hope to find a better method for treatment. This severe epilepsy syndrome is caused by genetic mutations. Patel is currently researching how gene therapy might be used to correct this variant. Instead of simply suppressing seizures, this method would address the root genetic cause. This would create a one-time curative treatment for one of the most harmful pediatric epilepsy variants, and potentially a model for more genetically-based epilepsies in the future.
You can read more of Patel's published research here.
Using Gene Therapy to Tackle Pediatric Epilepsy
I love my research because it's really exploring the unknown and I love to take that journey with the talented grad students, postdocs, and faculty that I have that have the single goal of better understanding epilepsy. Technologies have advanced so much that we have really clinically relevant models that we can use to better understand epilepsy. We can now take cells from these patients and grow them in the lab and make them into neurons so that we can really understand at the cellular level what's going on in these patients to generate seizures and cause epilepsy.
My name is Manoj Patel, and I'm a professor in the Department of Anesthesiology. And my research focuses on a rare pediatric epilepsy known as SCN8A epileptic encephalopathy.
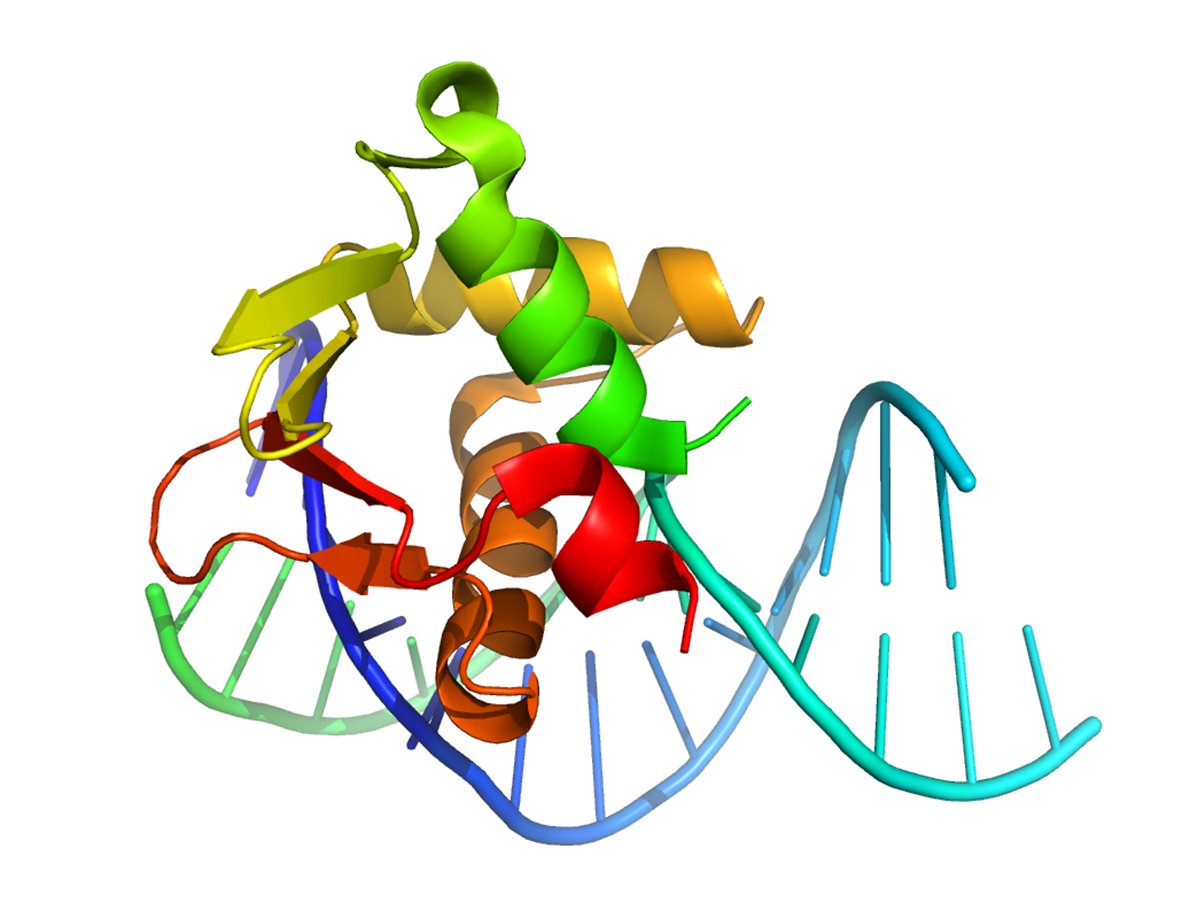
The SCN8A gene encodes for the Nav1.6 sodium channel isoform, which is heavily expressed in the brain. Mutations in this channel lead to aberrant activity and seizure generation. So we're really interested in understanding how a single base change in this protein can lead to epilepsy.
Patients with epilepsy are treated with anti-seizure medications that suppress the seizures. However, they don't go after the underlying mechanism for their epilepsy. With the help of very talented graduate students, we are using gene therapy techniques known as base editing. We are able to go in and correct the abnormal variant and make it into a healthy variant, thereby treating the underlying cause. This really could be a permanent fix for these patients.
What are you working on right now?
Our laboratory studies SCN8A-related developmental and epileptic encephalopathy (SCN8A-DEE), a severe genetic epilepsy caused by pathogenic variants in SCN8A, the gene encoding the neuronal sodium channel Nav1.6. Nav1.6 plays a central role in neuronal firing, and many disease-causing SCN8A variants result in a gain-of-function, leading to neuronal hyperexcitability. To date, more than 800 patients with de novo pathogenic SCN8A variants have been identified worldwide.
Clinically, individuals with SCN8A-DEE experience early-onset, treatment-resistant seizures and a broad range of comorbidities, including developmental delay, intellectual disability, movement disorders, and behavioral impairments. Despite aggressive anti-seizure therapy, seizure control is often poor, and the risk of sudden unexpected death in epilepsy (SUDEP) is substantially increased, underscoring the urgent need for disease-modifying treatments.
Because SCN8A-DEE is caused by a single, defined pathogenic variant, it represents an ideal candidate for precision gene therapy. We are developing a base editing-based gene correction strategy that directly targets and permanently corrects disease-causing SCN8A variants at the DNA level. Unlike conventional therapies that attempt to suppress seizures symptomatically, this approach addresses the root genetic cause of disease. If successful, this strategy has the potential to provide a one-time, curative treatment for patients with SCN8A-DEE and may serve as a model for treating other severe monogenic epilepsies.
What are the most intriguing potential clinical applications of your work?
Gene therapy approaches such as base editing have enormous therapeutic potential, not only for SCN8A-DEE, but also for a wide range of diseases caused by single-nucleotide pathogenic variants. By enabling precise, permanent modification of DNA, base editing offers the possibility of durable, one-time treatments rather than chronic disease management.
Importantly, the clinical utility of base editing extends beyond correction of pathogenic variants. This technology can also be used to therapeutically inactivate disease-relevant genes, even when no inherited mutation is present. A notable example is PCSK9, where base editing has been used to disrupt gene function as a treatment strategy for hypercholesterolemia, resulting in sustained reductions in LDL cholesterol levels. These advances highlight the broad clinical applicability of base editing as a transformative therapeutic platform.
What recent discovery has impacted the way you think?
Base editing technology was pioneered by Professor David R. Liu and colleagues at the Broad Institute, whose work led to the development of cytosine and adenine base editors. These tools enable precise, single-base pair changes in DNA without introducing double-strand breaks and have been instrumental in advancing the field of therapeutic genome editing. The discovery and refinement of base editors have laid the foundation for multiple emerging gene therapy strategies now entering preclinical and clinical development.
What made you choose UVA Health as the place to do your research?
I first came to the University of Virginia as a postdoctoral fellow and was immediately struck by the collaborative research culture and opportunities for meaningful scientific partnership. After completing my postdoctoral training, I continued my scientific development in the UK before returning to UVA to establish my independent research laboratory. Charlottesville’s strong sense of community and quality of life have also made it a wonderful place to raise a family.
What do you wish more people knew about your area of research?
SCN8A-DEE is a rare disorder, with an estimated incidence of approximately 1 in 52,000 individuals. As an orphan disease, therapeutic development has historically been limited. However, the clinical and scientific relevance of SCN8A extends well beyond this rare patient population.
The SCN8A gene encodes Nav1.6, a sodium channel that is upregulated in temporal lobe epilepsy and plays a critical role in enhancing neuronal excitability and facilitating seizure initiation. Consequently, elucidating the mechanisms of seizure generation in SCN8A-DEE has implications not only for affected patients but also for common forms of epilepsy, including acquired epilepsies. Insights gained from this monogenic disorder may therefore inform broader therapeutic strategies aimed at modulating neuronal excitability and seizure susceptibility across the epilepsy spectrum.
How did you become interested in your area of research?
I have long been interested in the brain and the mechanisms by which neuronal networks generate and regulate activity. My training in neuroscience and electrophysiology led me to focus on the role of ion channels — particularly voltage-gated sodium channels — in controlling neuronal excitability. Following the identification of the first patient-derived SCN8A pathogenic variant in 2012, my research interests increasingly aligned with this severe pediatric epilepsy.
Through direct interactions with families of children diagnosed with SCN8A-DEE, I gained firsthand insight into the profound clinical challenges associated with this disorder. The difficulty in achieving adequate seizure control with existing therapies has been a major motivating force behind my efforts to develop more effective, disease-modifying treatments, particularly gene therapy approaches that directly target the underlying genetic cause.